
What is the Unique Device Identifier (UDI)
Purpose of this Quick Start Guide (QSG)
Provides a foundational understanding of the U.S. FDA UDI Rule to facilitate not only regulatory compliance but also successful adoption to achieve benefits across clinical, supply chain and financial functions.
Terminology
See the Glossary Quick Start Guide for the definitions to any unfamiliar terms or acronyms.
The UDI Rule
Congress tasked the FDA with establishing the Unique Device Identifier (UDI) system to improve patient safety through greater visibility around medical devices. The rule (21 CFR 801.30) requiring medical device manufacturers to assign and label medical devices with UDIs was issued September 24, 2013, and the phase in period ended September 24, 2024. The rule also requires manufacturers to publish additional information about those products in the Global UDI Database (GUDID).
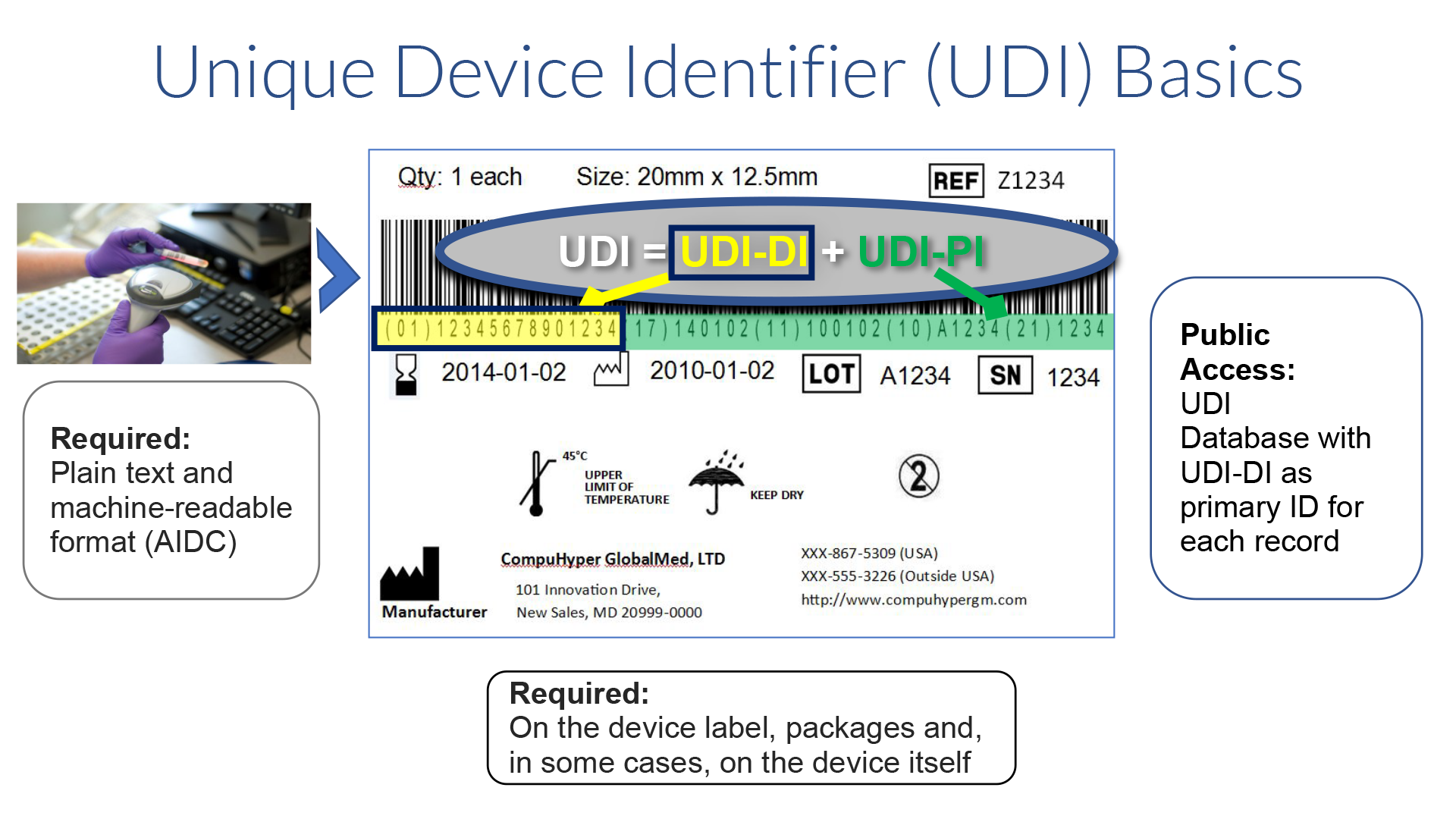
All medical devices (except those with a written exemption from the FDA) must have a UDI displayed on the label, package, and in some cases on the product itself, in human (text) and machine-readable (AIDC) formats.
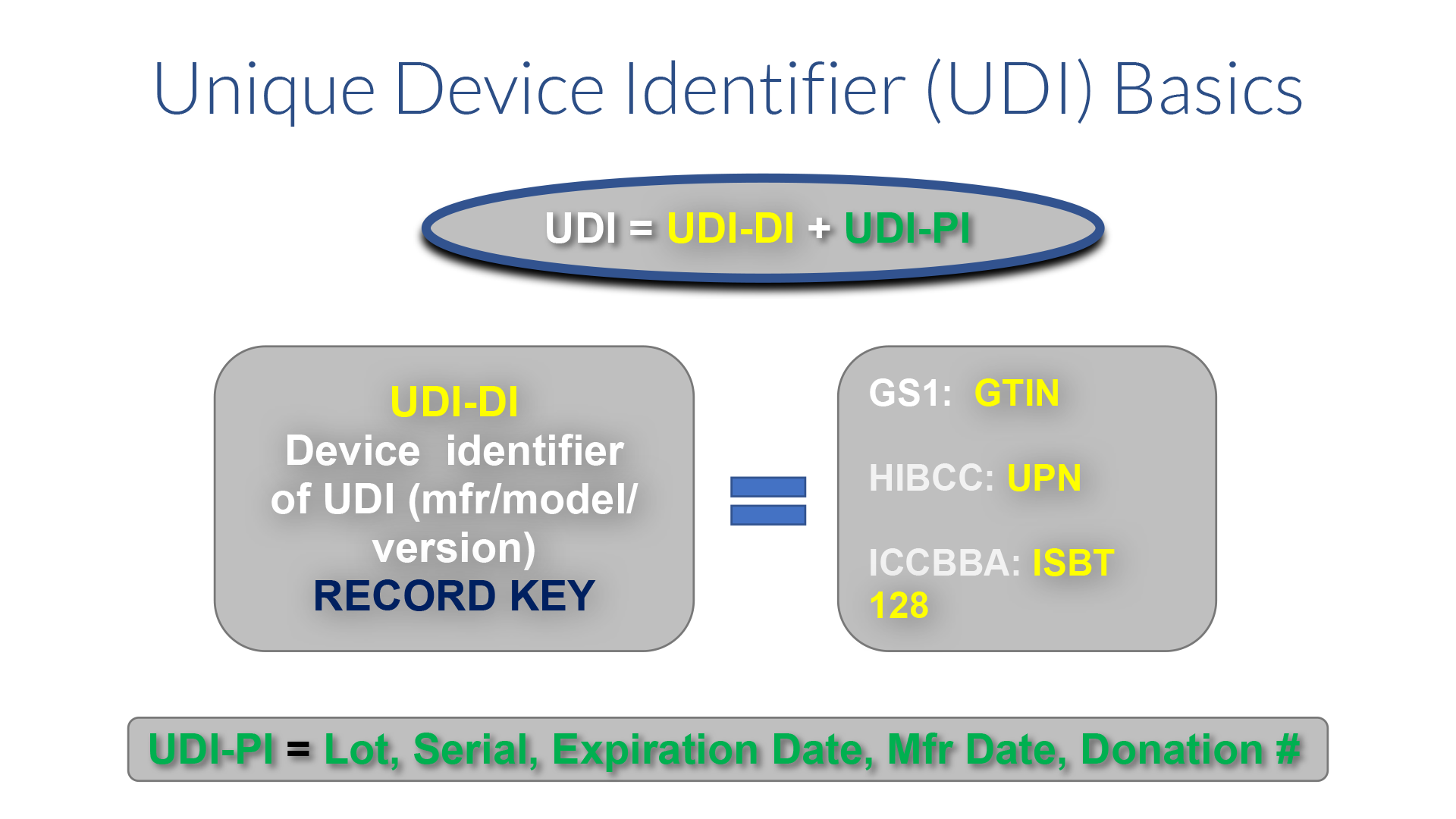
The FDA defines the UDI as a globally unique numeric or alphanumeric code that unambiguously identifies a medical device throughout its distribution and use. It is comprised of a mandatory, fixed Device Identifier (DI), which identifies the specific version or model and packaging quantity (unit of measure), and a variable Production Identifier (PI) that includes the information used by the manufacturer to control production, such as lot, batch, or serial number, expiration date and manufacture date. For a human cell, tissue, cellular, or tissue-based product (HCT/P) regulated as a medical device, the PI includes a distinct identification code that associates the HCT/P with the donor. If the label of a device is not required to have a particular PI, then it is not required to be included in the UDI. The manufacturer is responsible for obtaining UDIs from one of three approved issuing agencies: GS1, HIBCC, or for HCT/P, ICCBBA. Each agency has its own format and rules about how UDIs are allocated. Both the issuing agency and the FDA have rules dictating when a UDI-DI must be changed, (e.g., when a brand name changes or significant changes are made to the product itself). If there is a conflict, FDA regulations take precedent. Approximately 85% of the items in the GUDID contain UDIs issued by GS1.
The FDA requires specific data to be published to the GUDID, although manufacturers are also encouraged to include optional data elements, such as the catalog number. The UDI-DI is the key used to link this information to the device. Information in the GUDID is required to be updated any time there is a change to the UDI-DI or any of the other required data elements, (e.g. latex is removed from a medical device). If required data is not kept up-to-date in the GUDID, manufacturers are not allowed to sell those devices in the U.S. Please refer to the GUDID Data Elements Reference Table (DERT), available at www.fda.gov/udi.for more information on data elements contained in the GUDID. The majority of the information published in the GUDID is publicly available through AccessGUDID.
Under separate regulations from CMS and the ONC, health care providers are also required to use the UDI when documenting the use of implantable devices in electronic health records and in adverse event reports involving serious injury or death.
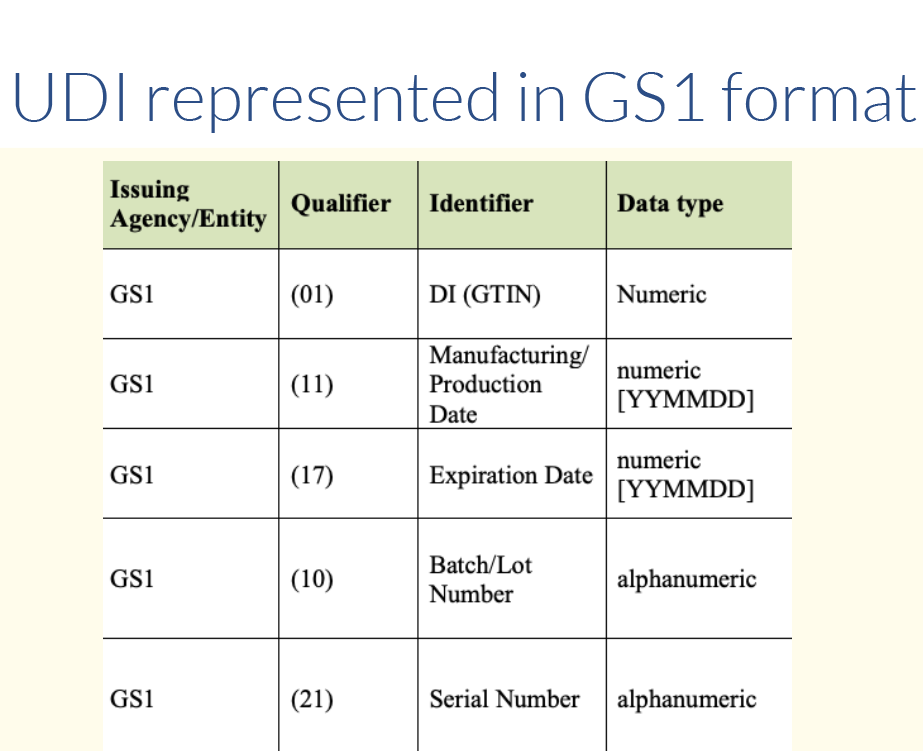
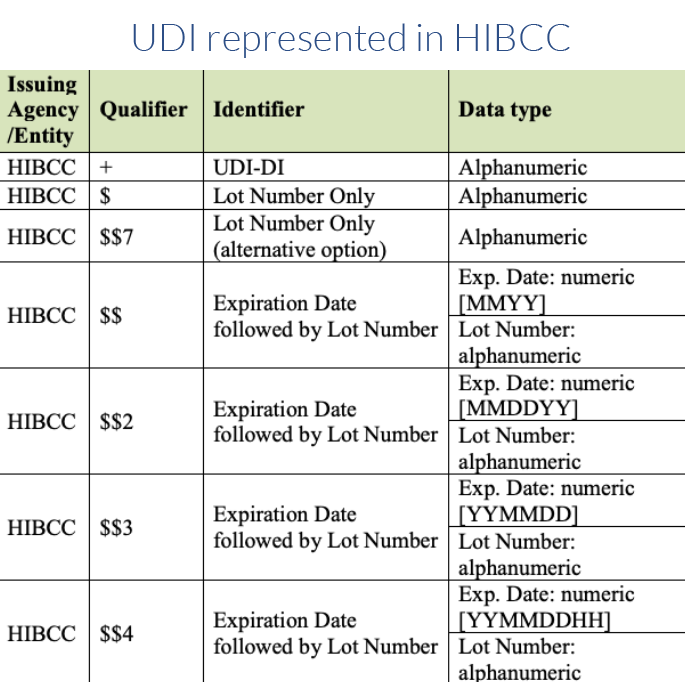
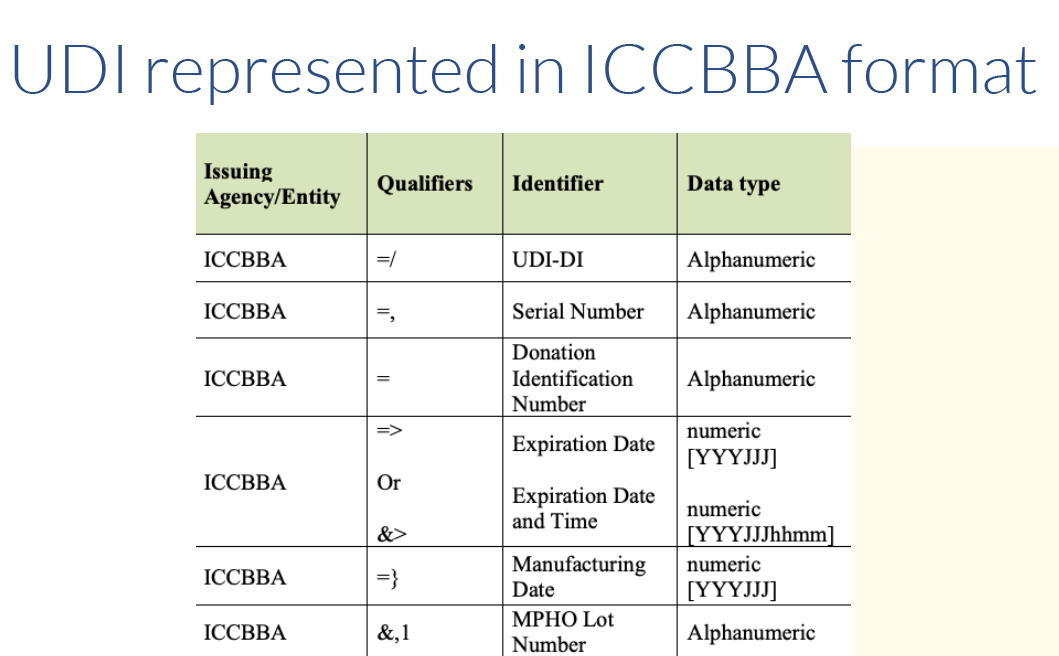
Issuing Agency UDI Formats and Qualifiers
Each issuing agency uses specific qualifiers that identify the type of information that follows in the body of the full UDI. These qualifiers can be used to determine which agency issued the UDI, as well as to help identify which barcode on a product contains the UDI. See the Quick Start Guide on Which Barcode to Scan.
RESOURCES
Easy, non-technical explanation of UDI components, labeling requirements, exceptions, and how UDI improves safety.
FDA UDI Rule, Guidance, Training, and Resources
Contains the full regulatory framework, compliance guides, and official training materials for UDI implementation.
21 CFR Part 830 – Unique Device Identification System Requirements
Establishes requirements for labeler responsibilities, issuing agencies, GUDID submission, and maintenance of UDI data.